

MỤC LỤC BÀI VIẾT
Giải trình tự gen 16S rRNA: Hành trình khám phá hệ vi sinh vật miệng từ A đến Z
Hệ vi sinh vật, một cộng đồng đa dạng các vi sinh vật sống trong và trên cơ thể chúng ta, đóng vai trò thiết yếu đối với sức khỏe con người. Đặc biệt, khoang miệng là một trong những hệ vi sinh vật phong phú và đa dạng nhất trong cơ thể người. Trung bình, một người có thể chứa từ khoảng 100 đến 300 loài vi khuẩn trong miệng, trong tổng số hơn 700 loài đã được phát hiện, cùng với nhiều loài vi khuẩn cổ, nấm, động vật nguyên sinh và virus. Mối quan hệ hai chiều giữa hệ vi sinh vật miệng và hệ gen người là cực kỳ quan trọng, không chỉ để duy trì sức khỏe mà còn liên quan đến sự phát triển của các bệnh lý. Trên thực tế, hệ vi sinh vật miệng đóng vai trò quan trọng trong sự khởi phát và tiến triển của hai bệnh phổ biến nhất ở người: sâu răng và viêm nha chu.
Trong những thập kỷ gần đây, những tiến bộ trong công nghệ giải trình tự thông lượng cao đã cho phép đặc trưng hóa hệ vi sinh vật miệng ở những mức độ sâu chưa từng có, điều mà các phương pháp trước đây không thể đạt được. Cụ thể, giải trình tự gen 16S ribosomal ribonucleic acid (rRNA) là một trong những kỹ thuật được sử dụng rộng rãi nhất để xác định sự đa dạng, cấu trúc và thành phần của các cộng đồng sinh vật nhân sơ liên quan đến sức khỏe và bệnh lý răng miệng. Kỹ thuật này vẫn được sử dụng phổ biến chủ yếu nhờ vào tốc độ xử lý nhanh, sự đơn giản trong phân tích kết quả và chi phí thấp.
Tuy nhiên, để có được kết quả nghiên cứu đáng tin cậy và sâu sắc bằng kỹ thuật giải trình tự gen 16S rRNA, các nhà nghiên cứu cần phải hiểu rõ và tối ưu hóa từng bước trong quy trình làm việc.
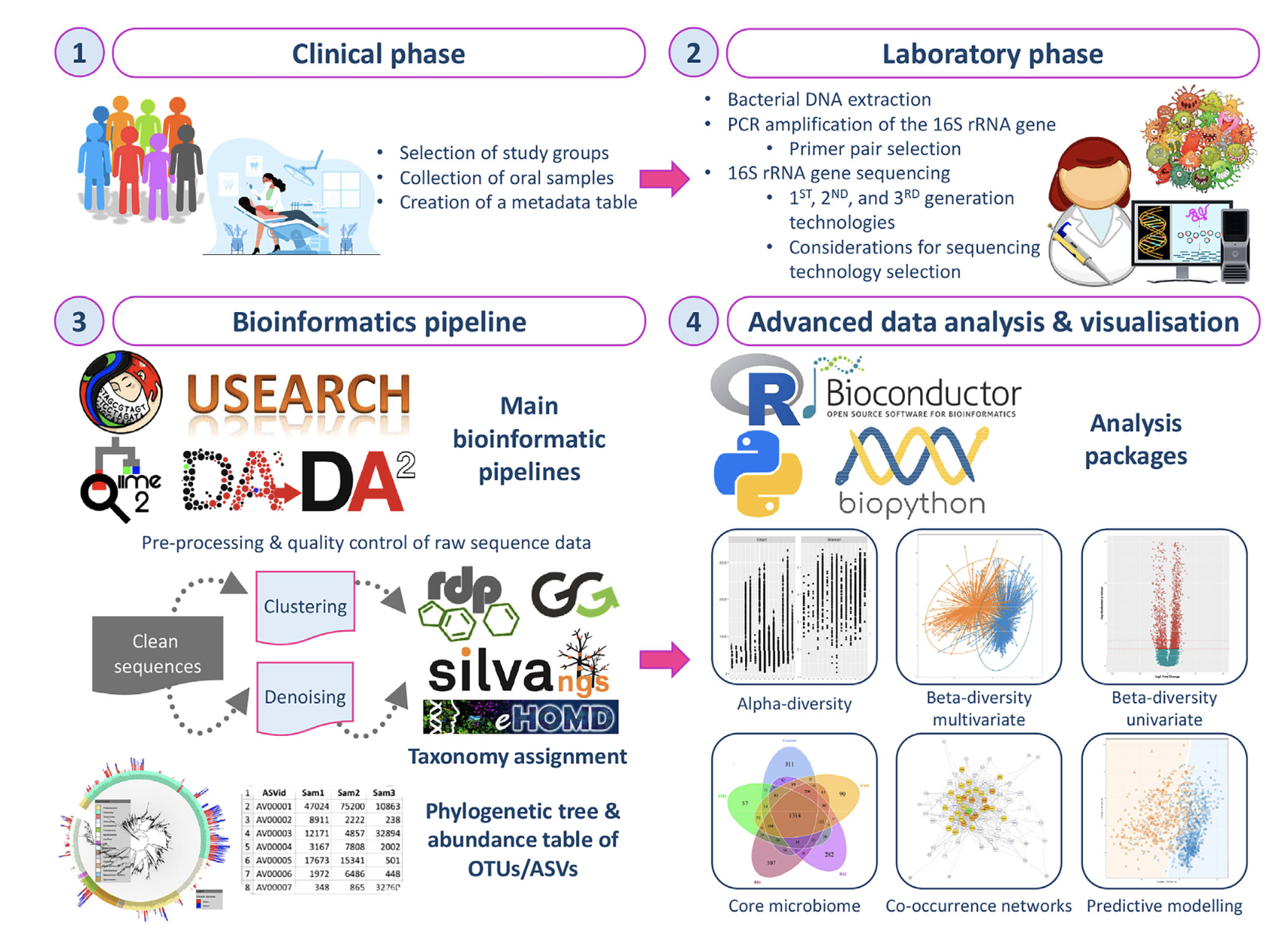
Gen 16S rRNA: Chỉ dấu phát sinh chủng loại đầy giá trị nhưng cũng có hạn chế
Gen 16S rRNA là đại phân tử được sử dụng rộng rãi nhất trong các nghiên cứu phát sinh chủng loại và phân loại sinh vật nhân sơ (vi khuẩn và vi khuẩn cổ). Gen này xen kẽ các vùng bảo tồn (conserved regions – C1-C10) chung cho tất cả các vi sinh vật, với các vùng biến đổi (variable regions – V1-V9). Các vùng bảo tồn rất hữu ích cho việc thiết kế các mồi (primer) cho phép khuếch đại các vùng siêu biến thiên (hypervariable regions). Gen 16S rRNA được coi là chỉ dấu phát sinh chủng loại chính vì một số lý do:
- Nó có mặt ở tất cả vi khuẩn và vi khuẩn cổ
- Nó thể hiện sự ổn định tương đối khi kết hợp các vùng bảo tồn và siêu biến thiên; kích thước tương đối lớn của gen làm cho nó phù hợp cho mục đích tin sinh học
- Sự bảo tồn trong cấu trúc bậc hai của nó tạo điều kiện cho sự xếp thẳng hàng (alignment) chính xác
- Sự dễ dàng trong việc giải trình tự gen này đã dẫn đến sự sẵn có của các cơ sở dữ liệu phong phú và không ngừng mở rộng
Tuy nhiên, việc sử dụng gen 16S rRNA cũng có những hạn chế. Một trong những hạn trọng nhất là tính dư thừa (redundancy) gen trong cùng một bộ gee. Khoảng 94% vi khuẩn và khoảng 53% vi khuẩn cổ trong miệng có nhiều hơn một bản sao gen 16S rRNA trong bộ gen của chúng. Điều này ảnh hưởng đến việc ước tính số lượng dựa trên số lượng bản sao gen, dẫn đến việc các taxa có số lượng bản sao thấp có xu hướng bị ước tính thấp hơn và ngược lại. Hơn nữa, nhiều bản sao gen trong cùng một bộ gen có thể khác nhau, với khoảng 66% vi khuẩn và khoảng 31% vi khuẩn cổ trong miệng có số lượng biến thể gen (sequence variants) trung bình lớn hơn 1. Sự biến đổi này có thể khiến các trình tự gen 16S khác nhau trong một bộ gen bị phân loại nhầm là thuộc về các taxa khác nhau. Mặc dù đã có các phương pháp để hiệu chỉnh sự biến đổi này, nhưng việc chuẩn hóa số lượng bản sao gen dường như không cải thiện được các phân tích giải trình tự gen 16S rRNA trong các tình huống thực tế.
Lựa chọn cặp mồi: Yếu tố then chốt ảnh hưởng đến kết quả
Để đạt được sự đa dạng tối đa khi nghiên cứu một hệ sinh thái, cặp mồi được chọn phải được tối ưu hóa phù hợp. Điều này bao gồm tối đa hóa hiệu quả và tính đặc hiệu cho mục tiêu khuếch đại (gen 16S rRNA), tối đa hóa phạm vi phát hiện (detection coverage) trong các mẫu, và tối đa hóa chiều dài của các đoạn khuếch đại (amplicons) được giải trình tự để cho phép xác định các cấp phân loại thấp hơn. Các nghiên cứu gần đây trong vi sinh vật học đã đánh giá nhiều cặp mồi và nhấn mạnh rằng việc lựa chọn cặp mồi ảnh hưởng đáng kể đến ước tính đa dạng và phân loại vi sinh vật.
Quy trình tin sinh học: Biến dữ liệu thô thành thông tin ý nghĩa
Một quy trình trong tin sinh học là một chuỗi các thuật toán được thực hiện theo một trình tự xác định trước để xử lý dữ liệu giải trình tự. Các pipeline phổ biến bao gồm mothur, USEARCH, dada2, QIIME và QIIME2. Mục tiêu chính của pipeline tin sinh học là thu được thông tin cần thiết cho phân tích sinh thống kê tiếp theo, bao gồm bảng đếm (count table), phân loại vi sinh vật và cây phát sinh chủng loại (phylogenetic tree). Các bước chính bao gồm:
1. Tách mẫu (Demultiplexing), tiền xử lý, lắp ráp contig (nếu cần) và kiểm soát chất lượng trình tự thô. Bước này bao gồm loại bỏ các đoạn đọc chất lượng thấp dựa trên Phred score và loại bỏ các lỗi do PCR hoặc giải trình tự
2. Thu thập trình tự duy nhất (unique sequences), số lượng và xếp thẳng hàng trình tự. Các đoạn đọc giống hệt nhau được kết hợp để giảm tải tính toán.
3. Phân cụm trình tự (Sequence clustering) hoặc khử nhiễu (denoising). Đây là bước quan trọng để nhóm các trình tự tương tự.
- Operational Taxonomic Units (OTUs) là các cụm sinh vật có sự tương đồng ở mức trình tự vượt qua một ngưỡng nhất định (thường lớn hơn 97% cho cấp loài). Phương pháp này có thể làm mất độ phân giải taxonomic nhưng tránh được vấn đề phân biệt biến thể sinh học và kỹ thuật.
- Amplicon Sequence Variants (ASVs) là các trình tự duy nhất ở độ phân giải nucleotide (100% tương đồng trình tự) được tạo ra bởi các thuật toán khử nhiễu. Phương pháp này cố gắng mô hình hóa lỗi của máy giải trình tự để phân biệt các biến thể sinh học thực sự với lỗi kỹ thuật. Các pipeline dựa trên ASV như dada2, Deblur, UNOISE thường cho thấy độ nhạy, đặc hiệu và độ chính xác cao hơn so với các thuật toán OTU.
4. Phân loại Taxonomic (Taxonomic assignment): Mỗi trình tự đại diện (từ OTU hoặc ASV) được gán một danh tính phân loại bằng cách so sánh với cơ sở dữ liệu tham chiếu. Các cơ sở dữ liệu phổ biến bao gồm RDP, Greengenes, SILVA và các cơ sở dữ liệu chuyên biệt cho môi trường trong miệng như CORE, HOMD, eHOMD. Việc sử dụng các cơ sở dữ liệu chuyên biệt cho môi trường nghiên cứu có thể giúp phân loại chính xác hơn. Sau khi có bảng đếm và phân loại taxonomic, cây phát sinh chủng loại được xây dựng để thể hiện mối quan hệ tiến hóa giữa các trình tự.
Phân tích dữ liệu nâng cao: Hiểu sâu về cộng đồng vi sinh vật
Hiểu được sự khác biệt về thành phần của cộng đồng vi sinh vật là điều cần thiết. Bảng OTU/ASV cùng với dữ liệu metadata (thông tin liên quan đến mẫu như điều kiện môi trường, loại mẫu, thời gian/địa điểm thu thập, sức khỏe vật chủ, v.v.) là cơ sở để thực hiện các phân tích nâng cao.
1. Chuẩn hóa dữ liệu (Data normalisation): Dữ liệu hệ vi sinh vật có đặc điểm thưa thớt (nhiều giá trị 0) và độ sâu giải trình tự không đồng đều giữa các mẫu. Ngoài ra, dữ liệu này có bản chất thành phần (compositional), nghĩa là số lượng đọc của một taxon phụ thuộc vào số lượng đọc của các taxon khác, và việc bỏ qua điều này có thể dẫn đến kết quả sai lệch. Các phương pháp chuẩn hóa truyền thống như tính toán độ phong phú tương đối (Total Sum Scaling – TSS) hoặc rarefaction (lấy mẫu con) có những hạn chế. Các phương pháp hiện đại hơn dựa trên biến đổi log-ratio (như ALR, CLR, ILR) được phát triển để xử lý tính chất thành phần của dữ liệu.
2. Tính toán hệ số đa dạng sinh học của cộng đồng vi sinh vật (Biodiversity of the microbial community):
- Đa dạng Alpha (Alpha-diversity) đo lường sự đa dạng trong một mẫu duy nhất. Các chỉ số phổ biến bao gồm độ phong phú (richness – số lượng taxa được phát hiện, như Observed richness, Chao1, ACE) và độ đồng đều (evenness – sự phân bố tương đối của các taxa, như Shannon-Weaver, Simpson, Pielou). Các chỉ số đa dạng alpha có tính đến cả khoảng cách phát sinh chủng loại (divergence-based) như Faith PD.
- Đa dạng Beta (Beta-diversity) đo lường sự đa dạng giữa nhiều mẫu. Nó mô tả mức độ chia sẻ taxa hoặc sự tương đồng giữa các cộng đồng. Các chỉ số truyền thống tập trung vào sự chồng lấp thành phần taxa (Jaccard, Bray-Curtis). Các chỉ số có thông tin phát sinh chủng loại (phylogenetically informed) tính đến mối quan hệ tiến hóa giữa các taxa, như UniFrac (unweighted và weighted). Các phương pháp phân tích đa biến (multivariate analysis) như PCA, PCoA, NMDS, t-SNE, RDA, GLM, Mantel test, ANOSIM, PERMANOVA, MRPP được sử dụng để đánh giá sự khác biệt giữa các cộng đồng dựa trên ma trận khoảng cách.
- Phân tích sự phong phú khác biệt (Analysis of differential abundances): Xác định các taxa có sự phong phú khác biệt đáng kể giữa các nhóm mẫu. Các phương pháp như Metastats, LEfSe, MaAsLin2, ANCOM, ANCOM-BC, LinDA đã được phát triển để đối phó với các thách thức của dữ liệu hệ vi sinh vật như tính thưa thớt và tính thành phần. Việc hiệu chỉnh cho kiểm định đa bội (multiple testing correction) là rất quan trọng.
3. Phân tích mạng vi sinh vật (Microbial network analysis): Khám phá các mẫu đồng xuất hiện (co-occurrence patterns) hoặc tương quan giữa các taxa trong hệ vi sinh vật. Mạng lưới bao gồm các “node” (OTU/ASV) và “edge” (mối quan hệ). Các chỉ số như mức độ node (node degree), độ trung tâm (node centrality – DC, CC, BC, EC) giúp mô tả cấu trúc mạng. Các phương pháp xây dựng mạng bao gồm kỹ thuật dựa trên tương quan (SparCC, SpiecEasi) và các phương pháp mới hơn có tính đến tính chất thành phần (SECOM)
4. Mô hình dự đoán (Predictive modelling): Sử dụng học máy (Machine Learning – ML) để xây dựng mô hình phân loại hoặc dự đoán dựa trên dữ liệu hệ vi sinh vật. Các phương pháp có thể là không giám sát (unsupervised – khám phá cấu trúc dữ liệu, như PCA, PCoA, t-SNE) hoặc có giám sát (supervised – phân loại mẫu vào các nhóm dựa trên dữ liệu đào tạo đã gán nhãn). Giảm hoặc lựa chọn biến (Variable reduction/selection) là bước quan trọng trước khi mô hình hóa, đặc biệt với dữ liệu chiều cao, để tránh đa cộng tuyến và cải thiện hiệu suất mô hình. Các kỹ thuật mô hình hóa phổ biến bao gồm SVM, sPLS-DA và các phương pháp mới phát triển cho dữ liệu hệ vi sinh vật như DMM, MetaPheno, Mdeep, MKMR. Điều cực kỳ quan trọng là tránh overfitting, tức là mô hình quá khớp với dữ liệu huấn luyện và không thể dự đoán tốt trên dữ liệu mới. Các kỹ thuật lấy mẫu lại (resampling) như k-fold cross-validation và Bootstrap là cách để đánh giá và tránh overfitting
Hiệu ứng Batch (Batch effects): Thách thức không thể tránh khỏi
Các nghiên cứu hệ vi sinh vật sử dụng giải trình tự gen 16S rRNA thường gặp phải kết quả mâu thuẫn. Một lý do chính cho sự bất đồng này là sự hiện diện của hiệu ứng batch (BEs) – bất kỳ nguồn biến đổi không mong muốn nào liên quan đến các yếu tố sinh học, kỹ thuật và tính toán không liên quan đến yếu tố sinh học quan tâm nhưng lại làm mờ nó đi. Các nguyên nhân có thể bao gồm sự khác biệt về đặc điểm vật chủ, giao thức thu thập/lưu trữ/xử lý mẫu (như tách DNA, PCR, giải trình tự) và các giao thức xử lý/phân tích dữ liệu. BEs gần như không thể tránh khỏi trong thực tế. Do đó, các phương pháp đã được phát triển để tính đến hoặc hiệu chỉnh chúng. Ban đầu, các công cụ này được phát triển cho dữ liệu microarray hoặc RNA sequencing (ví dụ: SVA, ComBat, removeBatchEffect) và yêu cầu biến đổi dữ liệu hệ vi sinh vật trước khi sử dụng. Gần đây, các phương pháp chuyên biệt cho dữ liệu hệ vi sinh vật đã xuất hiện (ví dụ: percentile normalisation, BDMMA, ConQuR, MMUPHin, các phương pháp dựa trên PLS-DA như PLSDA-batch). Tuy nhiên, các phương pháp này cũng có những hạn chế và hiệu quả của chúng cần được đánh giá thêm. Việc cân nhắc các sai lệch tiềm ẩn và nguyên nhân của chúng trong suốt quy trình làm việc giải trình tự gen 16S rRNA là rất quan trọng để có được kết quả đáng tin cậy.
Kết luận
Việc thực hiện một nghiên cứu hệ vi sinh vật bằng giải trình tự gen 16S rRNA đòi hỏi sự lựa chọn cẩn thận ở mỗi bước của quy trình làm việc. Từ việc hiểu rõ những hạn chế cố hữu của gen 16S rRNA và lựa chọn cặp mồi phù hợp, đến việc chọn công nghệ giải trình tự và độ sâu phù hợp. Việc lựa chọn pipeline tin sinh học và phương pháp xử lý dữ liệu (OTU vs. ASV) cũng có tác động đáng kể đến kết quả. Cuối cùng, các phân tích nâng cao (đa dạng alpha/beta, core microbiome, mạng lưới, mô hình dự đoán) và việc xử lý hiệu ứng batch là cần thiết để rút ra những kết luận có ý nghĩa lâm sàng. Trong tương lai, nghiên cứu nên tập trung vào các phương pháp phân tích chặt chẽ hơn, chẳng hạn như phát triển mô hình dự đoán để xác định các dấu ấn sinh học dựa trên hệ vi sinh vật nhằm phân loại trạng thái sức khỏe và bệnh tật. Việc hiểu rõ và kiểm soát các nguồn gây nhiễu, đặc biệt là hiệu ứng batch, sẽ là chìa khóa để đạt được kết quả tái lập và đáng tin cậy hơn.
LOBI Vietnam là công ty tiên phong trong lĩnh vực Đọc trình tự gen thế hệ mới NGS (Next Generation Sequencing) và Phân tích Tin sinh học. Liên hệ hotline/Zalo 092.510.8899 để biết thêm chi tiết.
