

MỤC LỤC BÀI VIẾT
Tổng quan và Đặt vấn đề
• Gen 16S rRNA từ lâu đã là công cụ chính để phân tích vi khuẩn dựa trên trình tự gen.
• Tuy nhiên, các kỹ thuật giải trình tự thông lượng cao trước đây (như Illumina) chỉ tạo ra các đoạn đọc ngắn, giới hạn việc giải trình tự ở một hoặc một vài vùng biến đổi (variable regions) của gen 16S thay vì toàn bộ gen (~1500 bp).
• Việc chỉ phân tích các vùng biến đổi này đã dẫn đến những giả định nhất định trong phân loại, ví dụ như ngưỡng tương đồng trình tự để xác định chi (>95%) hay loài (>97%). Mặc dù hữu ích, cách tiếp cận này bị hạn chế về độ phân giải taxonomic, thường chỉ đủ để xác định ở cấp độ chi trở lên.
• Mặt khác, các kỹ thuật ít thông lượng hơn đã sử dụng các điểm đa hình (polymorphisms), như SNP, trên gen 16S để phân biệt các chủng. Điều này cho thấy tiềm năng phân loại sâu hơn của gen 16S đầy đủ.
• Sự ra đời gần đây của các nền tảng giải trình tự thông lượng cao với đoạn đọc dài (long-read) như PacBio và Oxford Nanopore đã cho phép giải trình tự toàn bộ gen 16S một cách hiệu quả.
• Bài báo này đặt vấn đề cần đánh giá lại một cách nghiêm túc tiềm năng phân giải taxonomic ở cấp độ loài và chủng của kỹ thuật giải trình tự gen 16S, đặc biệt khi có thể giải trình tự toàn bộ gen và xử lý các biến thể trình tự.
Mô hình
Bài báo đã thực hiện một thí nghiệm in silico (trên máy tính) sử dụng các trình tự 16S đầy đủ từ các cơ sở dữ liệu công khai (như Greengenes).
• Họ đã cắt các trình tự đầy đủ này thành các đoạn “in-silico amplicons” tương ứng với các vùng biến đổi 16S thường được nhắm mục tiêu trong các nghiên cứu trước đây (ví dụ: V4, V1–V3).
• Bằng cách phân loại các đoạn amplicons này và phân cụm chúng thành OTUs ở các ngưỡng tương đồng khác nhau (97%, 98%, 99%), bài báo đã so sánh khả năng phân giải taxonomic của các vùng biến đổi so với gen 16S đầy đủ.
• Kết quả cho thấy:
- Các vùng biến đổi không thể đạt được độ chính xác phân loại ở cấp độ loài như khi sử dụng gen 16S đầy đủ. Vùng V4 cho kết quả kém nhất.
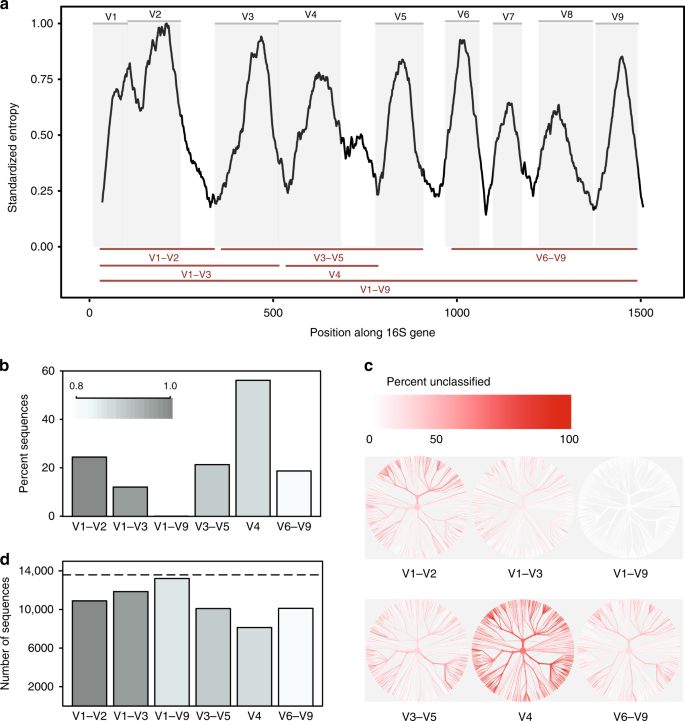
- Các vùng biến đổi khác nhau có xu hướng thiên lệch trong việc phân loại các nhóm vi khuẩn cụ thể (ví dụ: V1–V2 kém với Proteobacteria, V3–V5 kém với Actinobacteria).
- Việc chọn vùng biến đổi cũng ảnh hưởng đáng kể và không nhất quán đến số lượng OTUs được tạo ra khi phân cụm ở cùng một ngưỡng tương đồng.
• Kết luận từ thí nghiệm in silico là việc nhắm mục tiêu các vùng biến đổi chỉ đủ cho cấp độ chi trở lên và không đủ để phân biệt loài, đồng thời có thể làm ước tính thấp sự phong phú loài.
Ứng dụng
• Bài báo đã áp dụng kỹ thuật giải trình tự gen 16S đầy đủ sử dụng nền tảng đọc dài PacBio CCS trên các mẫu phân người và các vi khuẩn phân lập từ ruột người.
• Trên mẫu phân người, họ so sánh kết quả từ giải trình tự 16S đầy đủ (V1–V9), 16S một phần (V1–V3), và giải trình tự metagenomic toàn bộ bộ gen (mWGS).
• Phân tích chi Bacteroides cho thấy:
- Ước tính độ phong phú ở cấp độ chi là tương đương giữa 16S đầy đủ và 16S một phần.
- mWGS cho thấy sự đa dạng ở cấp độ loài lớn hơn.
- Giải trình tự 16S đầy đủ và phân cụm thành OTUs ở ngưỡng tương đồng 99% cung cấp ước tính hợp lý về độ phong phú của loài so với mWGS. Đáng chú ý, OTUs 99% này dường như gom nhóm tốt các bản sao gen 16S khác nhau trong cùng một bộ gen B. vulgatus.
- Họ cũng chứng minh rằng có thể phát hiện các biến thể nucleotide (đa hình) giữa các bản sao gen 16S trong cùng một bộ gen (B. vulgatus) ngay cả trong mẫu in vivo phức tạp.
• Trên 381 vi khuẩn phân lập từ ruột người, giải trình tự 16S đầy đủ cho thấy đa hình gen 16S trong cùng một bộ gen rất phổ biến (phát hiện SNP ở 349/381 chủng, tương ứng 54/61 OTUs 99%).
• Sự khác biệt trong hồ sơ biến thể nucleotide (SNP profiles) giữa các chủng phân lập được gán vào cùng một OTU 99% (cùng loài) cho thấy khả năng phân biệt ở cấp độ chủng. Các hồ sơ SNP này cung cấp một phương pháp mạnh mẽ để nhận dạng loài và phân biệt chủng.
Thảo luận và Hạn chế
Sự phát triển của các nền tảng đọc dài đã mở ra khả năng khai thác toàn bộ tiềm năng phân biệt của gen 16S.
• Tuy nhiên, bài báo cũng nhấn mạnh các hạn chế kỹ thuật của các nền tảng đọc dài hiện tại, đặc biệt là lỗi trình tự. Mặc dù PacBio CCS đủ chính xác để phát hiện các thay thế nucleotide (substitutions), lỗi chèn/xóa (insertions/deletions) vẫn là một vấn đề, đặc biệt là tại các đoạn lặp homopolymer.
• Họ đã chọn tập trung vào lỗi thay thế và bỏ qua lỗi chèn/xóa trong phân tích biến thể intragenomic, cho rằng đóng góp của lỗi chèn/xóa vào đa hình intragenomic có thể nhỏ hơn so với thay thế. Cần cải tiến công nghệ và thuật toán để xử lý tốt hơn các lỗi này trong tương lai.
• Một điểm thảo luận quan trọng là ý nghĩa của đa hình gen 16S trong cùng một bộ gen (intragenomic variation). Việc này có nghĩa là không thể đơn giản giả định rằng các đoạn đọc trình tự khác nhau một vài nucleotide (Exact Sequence Variants – ESVs) đại diện cho các taxa riêng biệt. Định lượng ESVs có thể làm ước tính quá mức sự phong phú của loài.
• Ngược lại, bài báo lập luận rằng khi được xử lý phù hợp, đa hình intragenomic này có thể làm tăng khả năng phân biệt giữa các taxa có quan hệ gần gũi. Việc phân cụm các biến thể trình tự gen 16S đầy đủ một cách phù hợp (ví dụ: OTUs 99%) có thể là một phương pháp giá trị để biểu diễn chính xác các loài.
Kết luận
• Giải trình tự các vùng biến đổi 16S là không đủ để đạt được độ phân giải taxonomic như khi giải trình tự toàn bộ gen 16S.
• Nền tảng giải trình tự đọc dài thông lượng cao hiện nay cho phép giải trình tự gen 16S đầy đủ với độ chính xác đủ để phát hiện các thay thế nucleotide giữa các bản sao gen 16S trong cùng một bộ gen.
• Đa hình gen 16S trong cùng một bộ gen là phổ biến trong các vi khuẩn ruột người và có thể được phát hiện chính xác bằng kỹ thuật giải trình tự đầy đủ.
• Mặc dù đa hình intragenomic làm phức tạp việc xử lý dữ liệu (ví dụ: không nên coi mỗi ESV là một loài riêng biệt), khi được xử lý đúng cách (ví dụ: phân cụm phù hợp như OTUs 99% trên trình tự đầy đủ), nó có tiềm năng làm tăng khả năng phân giải taxonomic.
• Bài báo kết luận rằng, với việc xử lý phù hợp dữ liệu trình tự 16S đầy đủ thông lượng cao, có thể đạt được khả năng phân loại chính xác các sinh vật riêng lẻ ở độ phân giải taxonomic rất cao, bao gồm cả cấp độ loài và chủng.
Tài liệu tham khảo: https://www.nature.com/articles/s41467-019-13036-1
LOBI Vietnam là công ty tiên phong trong lĩnh vực Đọc trình tự gen thế hệ mới NGS (Next Generation Sequencing) và Phân tích Tin sinh học. Liên hệ hotline/Zalo 092.510.8899 để biết thêm chi tiết.
