

TÓM TẮT
Giải trình tự amplicon gen ribosome RNA 16S (16S rRNA) bằng công nghệ giải trình tự thông lượng cao (HTS) hiện đang là phương pháp phổ biến nhất để khảo sát cộng đồng vi sinh vật đường ruột phức tạp. Tuy nhiên, kết quả nhận diện vi khuẩn có thể chịu ảnh hưởng đáng kể từ sự lựa chọn quy trình tin sinh học, dẫn đến thách thức trong việc so sánh giữa các nghiên cứu. Trong nghiên cứu này, chúng tôi tiến hành so sánh bốn quy trình phân tích phổ biến — QIIME2, Bioconductor, UPARSE và mothur — được thực thi trên hai hệ điều hành (Linux và macOS) nhằm đánh giá tác động của quy trình và hệ điều hành đến kết quả phân loại vi sinh vật trên 40 mẫu phân người. Tất cả các phân tích đều sử dụng cơ sở dữ liệu tham chiếu SILVA phiên bản 132.
Chúng tôi tiến hành so sánh độ chính xác phân loại và độ phong phú tương đối ở cấp độ ngành (phylum) và chi (genus) thông qua kiểm định tổng hạng Friedman. Kết quả cho thấy QIIME2 và Bioconductor tạo ra kết quả hoàn toàn tương đồng trên cả hai nền tảng hệ điều hành, trong khi UPARSE và mothur chỉ có sự sai khác tối thiểu. Việc gán nhãn phân loại các đơn vị phân loại (taxa) nhất quán giữa các quy trình, nhưng mức độ phong phú tương đối cho thấy sự khác biệt có ý nghĩa thống kê ở tất cả các ngành (p < 0.013) và phần lớn các chi phong phú nhất (p < 0.028), ví dụ như Bacteroides (QIIME2: 24.5%, Bioconductor: 24.6%, UPARSE-Linux: 23.6%, UPARSE-Mac: 20.6%, mothur-Linux: 22.2%, mothur-Mac: 21.6%, p < 0.001).
Những kết quả này chứng minh rằng quy trình tin sinh học có ảnh hưởng đáng kể đến việc ước lượng cấu trúc cộng đồng vi sinh vật, và do đó, các nghiên cứu sử dụng quy trình phân tích khác nhau không thể được so sánh trực tiếp một cách đáng tin cậy. Việc xây dựng một quy trình chuẩn hóa là điều cần thiết nhằm thúc đẩy sự phát triển ổn định và tái lập của lĩnh vực.
Từ khóa: Giải trình tự amplicon 16S rRNA; QIIME2; Bioconductor; UPARSE; mothur; hệ vi sinh vật đường ruột; mẫu phân người
GIỚI THIỆU
Hệ vi sinh vật đường ruột của con người (Gut Microbiota – GMB) hiện được xem là một lĩnh vực trọng điểm trong y học hiện đại, đóng vai trò thiết yếu trong việc làm sáng tỏ cơ chế bệnh sinh của nhiều bệnh lý mạn tính, cũng như hỗ trợ phát triển các liệu pháp điều trị mới. Các nghiên cứu gần đây đã chỉ ra rằng cộng đồng vi khuẩn cư trú trong đường tiêu hóa người có thể góp phần vào sự phát triển của nhiều bệnh khác nhau, bao gồm ung thư, rối loạn chuyển hóa, viêm mạn tính và thậm chí cả các rối loạn thần kinh.
Sự xuất hiện của công nghệ giải trình tự thông lượng cao (HTS) đã mở ra nhiều hướng tiếp cận mới cho việc phân tích hệ vi sinh vật, nhờ khả năng xác định các thành phần vi sinh với độ sâu và độ chính xác cao mà chi phí lại hợp lý. Một trong những phương pháp phổ biến là giải trình tự vùng biến thiên của gen 16S rRNA — một chỉ dấu phân tử quan trọng cho phép phân loại vi khuẩn và vi khuẩn cổ (archaea) dựa trên đặc điểm di truyền. Thông qua giải trình tự 16S rRNA, các nhà nghiên cứu có thể khảo sát được sự đa dạng và cấu trúc cộng đồng vi sinh vật trong hệ tiêu hóa, từ đó phát hiện các biến đổi tiềm ẩn liên quan đến bệnh lý.
Tuy nhiên, xử lý dữ liệu giải trình tự amplicon từ gen 16S rRNA là một quá trình phức tạp, đòi hỏi kỹ năng chuyên môn sâu trong lĩnh vực tin sinh học. Khối lượng dữ liệu lớn, sự không hoàn chỉnh của cơ sở dữ liệu nucleotide tham chiếu và sự đa dạng của các công cụ phân tích ở từng bước xử lý khiến quá trình phân tích trở nên thách thức với các nhà sinh học không chuyên về tin học. Thông thường, quá trình phân tích gồm ba bước chính: (i) ghép cặp các trình tự đọc (read pairs) thành đoạn đọc đơn dài hơn, (ii) kiểm soát chất lượng và cắt tỉa đoạn đọc, và (iii) gán nhãn phân loại học cho các trình tự. Mỗi bước có thể sử dụng nhiều thuật toán khác nhau và yêu cầu hiểu biết về cách thiết lập tham số hoặc kỹ năng lập trình, từ đó dẫn đến nhu cầu về hạ tầng tính toán và kiến thức chuyên môn đáng kể.
Trong những năm gần đây, nhiều quy trình phân tích được phát triển nhằm đơn giản hóa quy trình cho người dùng không chuyên, đồng thời nâng cao tính tái lập và minh bạch của phân tích. Trong số các quy trình phổ biến nhất hiện nay, QIIME2, Bioconductor, UPARSE và mothur là những đại diện tiêu biểu, cung cấp giải pháp trọn gói cho phân tích dữ liệu 16S rRNA từ thô đến kết quả phân loại học cuối cùng. Trong đó, QIIME2 và Bioconductor cho phép trích xuất các biến thể trình tự amplicon (ASVs), còn UPARSE và mothur sử dụng phương pháp phân cụm để tạo ra các đơn vị phân loại vận hành (OTUs).
Mặc dù đã có một số nghiên cứu đánh giá ảnh hưởng của quy trình tin sinh học đến kết quả phân loại vi sinh vật, phần lớn các nghiên cứu này sử dụng dữ liệu mô phỏng hoặc các cộng đồng mẫu chuẩn. Dữ liệu so sánh trực tiếp từ mẫu người thực tế vẫn còn hạn chế, đặc biệt thiếu vắng các nghiên cứu kết hợp đánh giá cả yếu tố hệ điều hành. Trong bối cảnh đó, nghiên cứu này được thiết kế nhằm đánh giá liệu lựa chọn quy trình tin sinh học và hệ điều hành có ảnh hưởng đến kết quả phân tích cộng đồng vi khuẩn trong mẫu phân người hay không. Chúng tôi sử dụng bốn quy trình chính (QIIME2, Bioconductor, UPARSE và mothur), thực thi trên hai nền tảng hệ điều hành (Linux và macOS), và phân tích sự khác biệt trong nhận diện vi sinh vật ở cấp độ ngành và chi cũng như độ phong phú tương đối của chúng.
Mẫu phân
Mẫu phân được thu thập từ các đối tượng tham gia trong một nghiên cứu đoàn hệ lớn hơn về lão hóa thần kinh, thực hiện tại 18 phòng khám chuyên khoa trí nhớ thuộc vùng Đông Lombardy, Ý. Nghiên cứu gốc — mang tên INDIA-FBP (Incremental Diagnostic Value of Florbetapir Amyloid Imaging) — nhằm đánh giá giá trị bổ sung của hình ảnh amyloid trong quy trình chẩn đoán lâm sàng đối với bệnh nhân có biểu hiện suy giảm nhận thức. Sau khi hoàn tất nghiên cứu gốc, 150 bệnh nhân và đối tượng đối chứng đã được mời cung cấp thêm mẫu phân và mẫu máu.
Theo quy trình đã công bố trước đó, mẫu phân được tự thu tại nhà trong cốc nhựa vô trùng, bảo quản ở -20°C, sau đó chuyển đến Viện IRCCS Fatebenefratelli tại Brescia trong vòng 24 giờ. Tại đây, mẫu được lưu trữ đông ở -20°C cho đến khi tiến hành phân tích. Trong nghiên cứu hiện tại, chúng tôi sử dụng mẫu từ một phân nhóm gồm 40 cá nhân có chức năng nhận thức dao động từ bình thường đến sa sút trí tuệ.
Tất cả các thủ tục nghiên cứu đã được Ủy ban Đạo đức của “Comitato Etico dell’IRCCS San Giovanni di Dio – Fatebenefratelli” (Brescia, Ý) phê duyệt (số hồ sơ 57/2014). Tất cả đối tượng tham gia đều đã ký văn bản đồng thuận sau khi được cung cấp đầy đủ thông tin.
Chiết xuất DNA
DNA tổng số được tách chiết từ 180–200 mg phân đông lạnh sử dụng bộ QIAamp DNA Stool Mini Kit (Qiagen, Đức) theo hướng dẫn của nhà sản xuất. Quá trình đồng nhất mẫu được thực hiện bằng kỹ thuật đánh bóng hạt (bead-beating) với thiết bị TissueLyser II trong 10 phút ở 30 Hz để đảm bảo phá vỡ cơ học cấu trúc mẫu. DNA sau khi chiết xuất được định lượng bằng thiết bị NanoDrop ND-1000 và lưu trữ ở +4°C để sử dụng sau.
Khuếch đại PCR, gắn chỉ thị và giải trình tự
Các vùng V3–V4 của gen 16S rRNA được khuếch đại theo giao thức chuẩn do Illumina đề xuất cho chuẩn bị thư viện metagenomic 16S. Sản phẩm amplicon được tinh sạch bằng hạt từ, rửa bằng ethanol 80%, sau đó hòa tan lại trong dung dịch Tris-HCl 10 mM. Tính toàn vẹn của amplicon được đánh giá bằng điện di trên gel agarose 2% có nhuộm ethidium bromide, trước khi lưu trữ ở -20°C.
Các amplicon sau đó được gắn chỉ thị kép bằng chỉ số Nextera XT theo quy trình chu trình nhiệt do nhà sản xuất khuyến nghị, tinh sạch và định lượng bằng phương pháp huỳnh quang (Qubit, Invitrogen). Độ dài fragment được xác định bằng chip Bioanalyzer DNA 1000 (Agilent), và thư viện cuối cùng được chuẩn hóa về nồng độ 4 nM, pha trộn, biến tính bằng NaOH 0.1N, pha loãng xuống 10 pM và nạp vào cartridge MiSeq v3 (Illumina) để tiến hành giải trình tự paired-end 2×300 chu kỳ. Các mẫu từ nhóm đối chứng và bệnh nhân sa sút trí tuệ được giải trình tự ở hai đợt riêng biệt.
[CHÈN HÌNH – Miêu tả quy trình chuẩn bị thư viện và giải trình tự]
Xử lý tin sinh học
Trình tự đọc 2×300 bp từ MiSeq được phân tích bằng bốn công cụ chính: QIIME2, Bioconductor, USEARCH, và mothur. Các phiên bản 64-bit được sử dụng cho QIIME2, Bioconductor và mothur; trong khi phiên bản 32-bit miễn phí của USEARCH (có giới hạn bộ nhớ) được sử dụng cho mục đích học thuật. Môi trường QIIME2 được thiết lập trong Conda.
Tất cả các công cụ được chạy trên hai nền tảng phần cứng giống nhau: một máy trạm Linux (Ubuntu 14.04.5, CPU Intel 8×3.70GHz, RAM 31.3GB) và một MacBook Pro (2018, macOS Mojave, CPU Intel Core i7 6 nhân 2.6GHz, RAM 16GB).
Với QIIME2, chúng tôi theo hướng dẫn “Moving Pictures”; với Bioconductor, sử dụng quy trình từ https://f1000research.com/articles/5-1492/v2; với USEARCH là tài liệu hướng dẫn chính thức của tác giả; còn mothur sử dụng giao thức MiSeq SOP trên trang chủ mothur.
Toàn bộ danh sách lệnh cụ thể được liệt kê trong Phụ lục 1–6.
PHÂN TÍCH DỮ LIỆU VÀ THỐNG KÊ
Tất cả phân tích thống kê và biểu đồ được thực hiện bằng phần mềm GraphPad Prism (phiên bản 8.1.1, GraphPad Software, San Diego, CA, Hoa Kỳ), ngoại trừ biểu đồ Venn được xây dựng bằng ứng dụng web jvenn.
Để đánh giá sự khác biệt giữa các quy trình tin sinh học, chúng tôi tiến hành so sánh các chỉ số sau:
- Số lượng trình tự đọc được giữ lại sau xử lý,
- Số lượng ngành (phylum) và chi (genus) được phân loại thành công,
- Độ phong phú tương đối của các đơn vị phân loại.
Cụ thể, kiểm định tổng hạng Friedman (Friedman rank sum test) với hiệu chỉnh Dunn được sử dụng để xác định sự khác biệt tổng thể giữa bốn quy trình. Trong khi đó, các so sánh cặp đôi giữa hai quy trình được thực hiện bằng kiểm định tổng hạng có dấu Wilcoxon (Wilcoxon signed-rank test).
Ngưỡng ý nghĩa thống kê được ấn định ở mức alpha = 0.05 cho tất cả các phân tích.
LƯU Ý KỸ THUẬT VỀ DỮ LIỆU
Dữ liệu đầu ra từ các quy trình phân tích đã được chuẩn hóa trước khi thống kê để đảm bảo tính tương thích giữa các định dạng. Các tham số cắt ngưỡng chất lượng (quality filtering), độ dài đoạn đọc sau trimming và loại bỏ chimera đều được giữ nhất quán theo khuyến nghị chính thức của từng công cụ.
Riêng với các quy trình sử dụng phương pháp ASV (QIIME2, Bioconductor), thuật toán DADA2 được triển khai để suy diễn chính xác biến thể trình tự sinh học từ dữ liệu thô. Với các quy trình OTU (UPARSE, mothur), cụm đại diện được xác định ở ngưỡng tương đồng 97% và 99%.
Cơ sở dữ liệu SILVA v132 và RDP v16 được sử dụng để gán nhãn phân loại học, tùy thuộc vào quy trình. Đối với UPARSE (không hỗ trợ trực tiếp SILVA), việc gán nhãn được thực hiện thông qua mothur sau khi xuất OTUs. Các trình tự đơn xuất hiện duy nhất trong một mẫu (singletons) đều bị loại bỏ trước phân tích thống kê.
KẾT QUẢ
Tác động của quy trình phân tích đến kết quả đầu ra
Tổng cộng 4.715.000 trình tự đọc thu được từ 40 mẫu phân đã được xử lý và phân tích bằng bốn quy trình tin sinh học: QIIME2, Bioconductor, UPARSE và mothur. Thời gian xử lý trung bình cho mỗi quy trình dao động từ chưa đến 1 giờ đối với UPARSE đến khoảng 9 giờ đối với mothur.
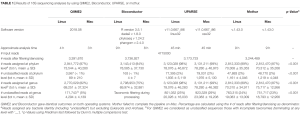
Kết quả cho thấy QIIME2 và Bioconductor tạo ra kết quả hoàn toàn tương đồng giữa hai hệ điều hành (Linux và macOS). Ngược lại, UPARSE và mothur cho thấy sự sai khác nhỏ, tuy không đáng kể về mặt thống kê (p > 0.999).
Sau bước lọc chất lượng và khử nhiễu, số lượng trình tự được giữ lại và gán nhãn trong QIIME2 và Bioconductor cao hơn rõ rệt so với hai quy trình còn lại:
- QIIME2: 3.391.670 đọc được giữ lại
- Bioconductor: 3.736.927
- UPARSE: 3.173.733 (bất kể hệ điều hành)
- mothur: 3.244.489 (bất kể hệ điều hành)
Tỷ lệ các đọc được gán nhãn ở cấp độ ngành dao động từ 84% đến 99%, trong khi ở cấp độ chi là từ 75% đến 99%, tùy theo quy trình. Đáng chú ý, UPARSE và mothur cho tỷ lệ cao hơn các trình tự không được phân loại ở cấp độ chi (từ 24–29%) so với QIIME2 (5%). Riêng Bioconductor loại bỏ trước các trình tự không phân loại như một bước mặc định.
Tác động đến phân loại học ở cấp độ ngành (phylum)
Tất cả quy trình đều xác định được các ngành vi khuẩn chính trong hệ vi sinh vật đường ruột người với sự đồng thuận cao. Cụ thể:
- Firmicutes: từ 46.6% (UPARSE-Mac) đến 48.3% (QIIME2)
- Bacteroidetes: từ 39.7% (QIIME2) đến 41.9% (UPARSE-Mac)
- Proteobacteria: từ 5.7% đến 6.0%
- Verrucomicrobia: 1.45% (UPARSE) đến 1.90% (Bioconductor)
- Actinobacteria: từ 1.44% đến 1.59%
- Tenericutes: từ 0.22% đến 0.34%
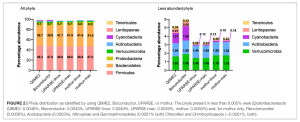
Mặc dù mức độ phong phú tuyệt đối có sự khác biệt nhỏ, phân tích thống kê cho thấy sự khác biệt có ý nghĩa thống kê giữa các quy trình (p < 0.0002). Đặc biệt, Lentisphaerae chỉ được phát hiện bởi QIIME2 và Bioconductor.
Tác động đến phân loại học ở cấp độ chi (genus)
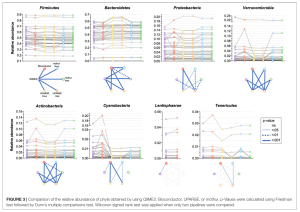
Ở cấp độ chi, QIIME2 và Bioconductor cho kết quả phong phú hơn đáng kể so với UPARSE và mothur (p < 0.001):
- QIIME2: 187 chi
- Bioconductor: 232 chi
- UPARSE-Linux: 120; Mac: 118
- mothur-Linux: 139; Mac: 138
Tổng cộng có 316 chi được xác định, trong đó chỉ có 78 chi xuất hiện đồng thời ở tất cả các quy trình. Bioconductor và QIIME2 chia sẻ nhiều chi nhất (147 chi), trong khi UPARSE-Mac và QIIME2 chia sẻ ít nhất (78 chi).
Sau khi lọc bỏ các chi có <5 đối tượng hoặc <10 đọc toàn dataset, xu hướng vẫn giữ nguyên. ( với 4A là trước khi lọc bỏ các chi, 4B là sau khi lọc bỏ )
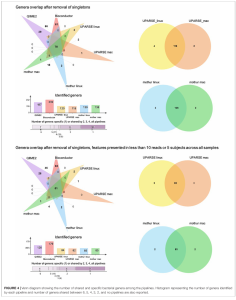
So sánh chi phong phú nhất
Chúng tôi chọn ra 10 chi có độ phong phú cao nhất từ mỗi quy trình để phân tích định lượng. Các chi như Bacteroides, Faecalibacterium, Alistipes và Subdoligranulum được phát hiện đồng thuận bởi cả bốn quy trình. Một số khác như CAG-352, Ruminococcus_1, Parabacteroides, Barnesiella và Prevotella_7 có sự khác biệt phụ thuộc vào công cụ phân tích.
[CHÈN HÌNH 5 – So sánh độ phong phú của 10 chi phổ biến nhất]
Phân tích thống kê cho thấy hầu hết các chi đều có sự khác biệt rõ rệt giữa các quy trình (p < 0.041), ngoại trừ Ruminococcus_1 và Ruminococcaceae (p > 0.074).
THẢO LUẬN
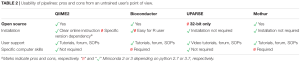
Trong bài báo này, chúng tôi đã so sánh bốn quy trình được sử dụng phổ biến là QIIME2, Bioconductor, UPARSE và mothur, chạy trên cả Linux và Mac OS, để đánh giá mức độ ảnh hưởng của các quy trình tin sinh học và hệ điều hành khác nhau đến việc phân loại học. Về tính dễ sử dụng của mỗi quy trình chúng tôi thấy rằng những quy trình được triển khai trong QIIME2 và UPARSE là thân thiện với người dùng nhất, vì chúng chỉ bao gồm một số lượng lệnh giới hạn với cú pháp đơn giản và yêu cầu kiến thức lập trình tối thiểu. Ngược lại, Bioconductor sử dụng ngôn ngữ R, cung cấp một giao diện thuận lợi cho người dùng đã được đào tạo và thường quen thuộc với R. Tuy nhiên, tài liệu chi tiết và phong phú của Bioconductor và mothur tạo điều kiện cho việc áp dụng chúng đối với người dùng có kinh nghiệm sử dụng dòng lệnh ở mức trung bình. Trong trường hợp của UPARSE, điều này đúng ngoại trừ bước gán nhãn phân loại học, vì quy trình UPARSE không cung cấp chức năng gán nhãn phân loại học cho các trình tự đại diện của OTU hoặc khuyến nghị về cách thực hiện nó. Sự khác biệt chính giữa các quy trình nằm ở thủ tục cài đặt. Thật vậy, USEARCH và mothur chỉ yêu cầu tải xuống một tệp thực thi, trong khi QIIME2 và Bioconductor yêu cầu cấu hình và cài đặt. Đặc biệt, như đã được ghi nhận trước đây trong một nghiên cứu tương tự sử dụng QIIME1, QIIME2 phụ thuộc vào một số chương trình, khiến việc cài đặt mất nhiều thời gian và công sức hơn.
Chúng tôi đã chỉ ra rằng các kết quả đầu ra khác biệt đáng kể giữa các quy trình mặc dù sử dụng cùng một dữ liệu đầu vào. Cụ thể, khi phương pháp phân cụm được sử dụng (ví dụ: phân cụm OTU 97% bằng UPARSE), kết quả là có ít chi riêng biệt hơn. Điều này có thể là do bước phân cụm đã gom nhầm các chi khác biệt lại với nhau do sự tương đồng trình tự (điều này sẽ chỉ dẫn đến việc một chi duy nhất được gán cho OTU), tức là việc phân cụm đã đánh giá thấp số lượng chi chính xác, hoặc do ASVs đôi khi bị gán sai, dẫn đến kết quả dương tính giả trong danh sách các chi được xác định. Chúng tôi cũng nhận thấy rằng, mặc dù cả hai phương pháp đều không sử dụng phân cụm, việc xử lý bằng Bioconductor dẫn đến số lượng chi được phát hiện nhiều hơn đáng kể so với QIIME2. Những ví dụ như vậy làm nổi bật thực tế rằng các thước đo độ phong phú của cộng đồng vi khuẩn (ví dụ: alpha- và beta-diversity) không thể được so sánh một cách đáng tin cậy cho các mẫu được xử lý bằng các quy trình khác nhau. Ví dụ, việc các quy trình khác nhau tạo ra số lượng sinh vật khác nhau sẽ dẫn đến các giá trị alpha-diversity khác nhau cho cùng một mẫu. Việc thực hiện các so sánh này chỉ bằng cách sử dụng các giá trị mặc định của quy trình sẽ dẫn đến sự khác biệt rõ ràng giữa các quy trình mà rất khó để giải thích, cả về chỉ số alpha- và beta-diversity

Chúng tôi cũng nhận thấy rằng nhiều đoạn đọc không được gán nhãn khi sử dụng SILVA với UPARSE và mothur (tạo ra OTUs) hơn so với QIIME2 (tạo ra ASVs). Phân cụm OTU nói chung tạo ra một trình tự đại diện (tức là, tâm của cụm) được chú thích về mặt phân loại học. Do việc xếp hạng các trình tự 16S rRNA duy nhất theo độ phong phú là bước đầu tiên của quy trình phân cụm trong UPARSE, điều này được cho là mang lại các trình tự đại diện có ý nghĩa sinh học hơn vì tâm của cụm luôn là trình tự phong phú nhất trong cụm. Ngược lại, ASVs có thể cho phép phát hiện các sinh vật quan trọng và khác biệt có khả năng có tỷ lệ hiện diện thấp trên các mẫu. Về mặt lý thuyết, điều này có thể phải trả giá bằng khả năng chú thích phân loại học cho chúng, nhưng dữ liệu của chúng tôi không chỉ ra điều này. Tuy nhiên, vì phương pháp tạo ASV khác được sử dụng (Bioconductor) đã loại bỏ các đoạn đọc chưa được gán nhãn trước khi phân tích, nên không rõ mức độ khác biệt này là do việc gán nhãn cho ASVs và OTUs hay do sự khác biệt trong quy trình.
Điều đáng ngạc nhiên đối với chúng tôi là việc áp dụng cùng các lệnh UPARSE và mothur (xem Phương pháp Bổ sung 3, 4) trên hai hệ điều hành khác nhau (Linux và Mac) lại tạo ra các kết quả khác nhau, và chúng tôi không thể xác định được lý do cho sự khác biệt quan sát được này.
Một điểm đáng chú ý là sự khác biệt rõ rệt nhất về độ phong phú tương đối giữa các quy trình được quan sát thấy ở các sinh vật có độ phong phú thấp. Ví dụ, Lentisphaerae chỉ được phát hiện bằng QIIME2 và Bioconductor (với độ phong phú tương đối dưới 1% trong cả hai trường hợp). Vấn đề này càng phức tạp hơn bởi thực tế là các sinh vật có độ phong phú thấp có khả năng ít được đại diện trong các cơ sở dữ liệu tham chiếu. Các quan sát tương tự có thể được rút ra từ một phân tích trước đó so sánh các quy trình MG-RAST và QIIME1, nơi các sinh vật có độ phong phú thấp có thể khác nhau về độ phong phú ước tính của chúng lên đến vài bậc độ lớn. Điều này có ý nghĩa vì có khả năng những sinh vật này vẫn đóng vai trò quan trọng trong các bệnh ở người.
Điều đáng lưu ý là có những sai lệch rõ ràng theo từng ngành cụ thể trong một số quy trình nhất định: đặc biệt, chúng tôi đã quan sát thấy sự khác biệt có ý nghĩa thống kê giữa QIIME2 và tất cả các quy trình khác trong việc chú thích ngành Firmicutes, và giữa QIIME2 và tất cả các quy trình khác ngoại trừ Bioconductor đối với ngành Bacteroidetes. Do đó, các quy trình xử lý khác nhau có thể dẫn đến sự khác biệt ngay cả ở cấp độ ngành, bao gồm hai ngành phong phú và phổ biến nhất của hệ vi sinh vật đường ruột người.
Một khó khăn mà chúng tôi xác định trong phân tích của mình là một số chi có sự đồng thuận tốt hơn giữa các quy trình so với các chi khác. Ví dụ, độ phong phú của chi Faecalibacterium có sự đồng thuận chung tốt giữa các kết quả từ tất cả các quy trình, trong khi Bacteroides thì kém hơn. Mặc dù chúng tôi không thể xác định lý do đằng sau những khác biệt này, chúng tôi có thể gợi ý rằng chúng có thể liên quan đến sự đa dạng trong các chi này: Faecalibacterium bao gồm ít loài có ít họ hàng gần, so với Bacteroides có sự đa dạng ở cấp độ loài rất lớn trong hệ vi sinh vật đường ruột người và do đó nhạy cảm hơn với các bước tin sinh học trước khi gán nhãn phân loại học. Hơn nữa, có thể việc lựa chọn phương pháp phân loại học (được tích hợp trong các cơ sở dữ liệu khác nhau) có thể ảnh hưởng đến kết quả một cách khác nhau ở các vùng khác nhau của cây phát sinh loài.
Nhìn chung, chúng tôi báo cáo các sự khác biệt liên quan đến việc xử lý cùng một dữ liệu thô bằng cách sử dụng các quy trình tin sinh học khác nhau. Mức độ của sự khác biệt này có thể so sánh với những gì đã được báo cáo bởi các nghiên cứu trước đây điều tra tác động của việc xử lý mẫu ở giai đoạn đầu (upstream sample processing). Các phương pháp chiết xuất DNA và các phòng thí nghiệm khác nhau đã được chứng minh là dẫn đến sự khác biệt lên đến 10 lần và 2 lần tương ứng về độ phong phú tương đối của các chi vi khuẩn cụ thể. Ví dụ, sự khác biệt về độ phong phú tương đối của chi Bacteroides có độ phong phú cao có thể chênh lệch hơn hai lần tùy thuộc vào phương pháp chiết xuất được sử dụng, và chúng tôi đã quan sát thấy sự khác biệt về độ lớn tương tự từ cùng một mẫu giữa các quy trình khác nhau hoặc thậm chí cùng một quy trình trên các hệ điều hành khác nhau. Tương tự, so sánh giữa nền tảng giải trình tự Illumina và 454 Titanium dẫn đến sự khác biệt lên đến hai lần về số lượng chi vi khuẩn được phát hiện, điều này có thể so sánh với sự khác biệt giữa Bioconductor và UPARSE hoặc mothur. Ngoài ra, một nghiên cứu điều tra các phương pháp thu thập và lưu trữ mẫu khác nhau đã báo cáo những khác biệt tương tự (mặc dù tương đối nhỏ) về độ phong phú tương đối của các ngành vi khuẩn chiếm ưu thế.
KẾT LUẬN
Ngoài sự khác biệt về thời gian chạy, tính dễ sử dụng và cài đặt, chúng tôi đã chứng minh rằng việc sử dụng các quy trình tin sinh học khác nhau có tác động mạnh mẽ đến các phân tích kết quả, với mức độ có thể so sánh với sự khác biệt trong việc xử lý mẫu ở giai đoạn đầu và các quy trình giải trình tự. Những khác biệt này bao gồm sự hiện diện của số lượng đọc được gán khác nhau, số lượng đơn vị phân loại riêng biệt được phát hiện trong tập dữ liệu, và độ phong phú tương đối của các sinh vật khác nhau trong cộng đồng vi khuẩn đường ruột. Tổng hợp lại, dữ liệu của chúng tôi chỉ ra rằng kết quả không thể so sánh được nếu thu được bằng cách áp dụng các quy trình khác nhau, và việc hài hòa hóa là hết sức cấp thiết để thúc đẩy lĩnh vực này phát triển. Như đã được lưu ý trước đây trong các tài liệu, việc tuân thủ các hướng dẫn hài hòa hóa cụ thể sẽ giảm thiểu đáng kể những khó khăn này. Các hướng dẫn như vậy nên báo cáo chính xác phiên bản phần mềm được sử dụng trong phân tích, chỉ rõ các tham số cụ thể đã được sử dụng (ngay cả khi đó là cài đặt mặc định), và tiêu chuẩn hóa các phương pháp cũng như bộ tham số trong và giữa các nhóm nghiên cứu. Ngoài ra, lĩnh vực này có thể sẽ được hưởng lợi từ việc làm việc càng nhiều càng tốt với các quy trình và khung làm việc hợp tác, mã nguồn mở như QIIME2, vốn tích hợp và liên tục được cập nhật với các phương pháp tiên tiến nhất được phát triển trong lĩnh vực. Các khung làm việc này sau đó có thể được tự động áp dụng cho bộ dữ liệu thô hiện có được lưu trữ trong các kho lưu trữ công cộng. Điều này có thể đòi hỏi một sự thay đổi khỏi việc nhấn mạnh vào các kết quả tĩnh trong một định dạng xuất bản không thể thay đổi, và hướng tới các cơ sở dữ liệu được cập nhật liên tục và công khai, chứa dữ liệu ở cả dạng thô và đã xử lý, chẳng hạn như MG-RAST.
LOBI Vietnam là công ty tiên phong trong lĩnh vực Đọc trình tự gen thế hệ mới NGS (Next Generation Sequencing) và Phân tích Tin sinh học. Liên hệ hotline/Zalo 092.510.8899 để biết thêm chi tiết.
