

Phân tích đánh giá toàn diện và không thiên vị về tám thuật toán xử lý dữ liệu trình tự 16S rRNA amplicon, bao gồm cả các phương pháp dựa trên Đơn vị Phân loại Thực nghiệm (OTUs) và Biến thể Chuỗi Amplicon (ASVs). Các nhà nghiên cứu đã sử dụng cộng đồng mock phức tạp nhất hiện có, gồm 227 chủng vi khuẩn, và cơ sở dữ liệu Mockrobiota để so sánh các thuật toán một cách khách quan, nhờ vào các bước tiền xử lý thống nhất.
Các kết quả chính cho thấy:
- Các thuật toán ASV, đặc biệt là DADA2, cho ra kết quả nhất quán nhưng lại mắc lỗi quá phân tách (over-splitting), nghĩa là một chuỗi tham chiếu được chia thành nhiều ASV.
- Các thuật toán OTU, dẫn đầu bởi UPARSE, tạo ra các cụm với ít lỗi hơn nhưng lại mắc lỗi quá gộp (over-merging), nghĩa là nhiều chuỗi tham chiếu được gộp vào cùng một OTU.
- Đáng chú ý, UPARSE và DADA2 cho thấy sự tương đồng gần nhất với cộng đồng vi sinh vật dự kiến về mặt thành phần và phân tích đa dạng alpha và beta.
Nghiên cứu này cung cấp một khung khổ vững chắc để so sánh các thuật toán OTU/ASV hiện có và đánh giá các công cụ mới một cách khách quan.
Trình tự 16S rRNA amplicon là một công cụ thiết yếu trong nghiên cứu hệ vi sinh vật để xác định thành phần vi khuẩn và độ phong phú tương đối của chúng trong một mẫu. Tuy nhiên, phương pháp này dễ mắc phải các sai lệch và lỗi kỹ thuật phát sinh trong quá trình thực hiện, như lỗi PCR, chuỗi giả (chimeric artificial sequences) và lỗi trình tự (sequencing errors). Những lỗi này làm ảnh hưởng nghiêm trọng đến ước tính đa dạng và gây khó khăn trong việc xác định các chuỗi sinh học thực sự đại diện cho cộng đồng vi sinh vật.
Để khắc phục nhiễu trình tự, phương pháp truyền thống là gộp các chuỗi dựa trên độ giống nhau (thường là 97%) vào một đơn vị phân loại duy nhất, gọi là OTU. Các phương pháp dựa trên gộp cụm bao gồm UPARSE, VSEARCH-DGC (sử dụng thuật toán gộp cụm tham lam), và mothur (tính toán ma trận khoảng cách và gộp cụm bằng các phương pháp nearest, furthest, hoặc average neighbor; cũng như Opticlust mới hơn). Mặc dù có những tiến bộ, việc sử dụng ngưỡng gộp cụm cố định (ví dụ 3% khác biệt) vẫn còn hạn chế, vì ngưỡng tối ưu nên phụ thuộc vào vùng gen và loại vi khuẩn.
Để giải quyết vấn đề này, các phương pháp gần đây đã phát triển Biến thể Chuỗi Amplicon (ASV), còn được gọi là Exact Sequence Variants (ESVs) hoặc zero radius OTUs (zOTUs). Các kỹ thuật này sử dụng các mô hình thống kê để phân biệt chuỗi thực với chuỗi giả. Các công cụ như DADA2, MED, UNOISE3 và Deblur áp dụng cách tiếp cận này. Ưu điểm rõ ràng của ASV là tính nhất quán, cho phép sử dụng chúng làm nhãn chuỗi trên các nghiên cứu khác nhau mà không cần gộp cụm lại. Tuy nhiên, phương pháp này cũng có thể tạo ra nhiều ASV cho các bản sao gen 16S rRNA không giống hệt nhau trong cùng một chủng.
Thách thức lớn trong việc so sánh các thuật toán này là sự đa dạng trong các thiết lập thử nghiệm, tiêu chí lọc và phương pháp loại bỏ chuỗi giả, làm che khuất sự hiểu biết rõ ràng về hiệu suất của chúng. Ngoài ra, việc thiếu “sự thật cơ bản” (ground truth) trong các mẫu cộng đồng vi sinh vật tự nhiên cũng gây khó khăn cho việc đánh giá chính xác. Các cộng đồng mock (mô phỏng) được sử dụng làm giải pháp thay thế, nhưng các bộ dữ liệu mock hiện có thường không đủ phức tạp để phản ánh sự đa dạng tự nhiên. Do đó, cần một phân tích so sánh khách quan sử dụng một mẫu mock phức tạp hơn để đánh giá độc lập điểm mạnh và hạn chế của các phương pháp gộp cụm/khử nhiễu.
Mục tiêu của nghiên cứu này là tiến hành một so sánh đối đầu không thiên vị và đầy thách thức giữa các phương pháp gộp cụm và khử nhiễu, làm nổi bật điểm mạnh và hạn chế của từng phương pháp. Đặc biệt, nghiên cứu khám phá sự khác biệt của chúng trong việc dự đoán thành phần cộng đồng mock, tỷ lệ lỗi, hành vi gộp/phân tách, phân tích đa dạng và thời gian chạy.
Phương pháp
Nghiên cứu đã sử dụng hai nguồn dữ liệu chính để đánh giá hiệu suất của các thuật toán:
- Dữ liệu mock HC227_V3V4: Được tạo ra từ cộng đồng mock HC227, bao gồm DNA bộ gen của 227 chủng vi khuẩn thuộc 197 loài khác nhau, nhắm mục tiêu vùng V3-V4 của gen 16S rRNA. Trình tự được thu thập trên nền tảng Illumina MiSeq4000.
- Dữ liệu từ Mockrobiota database: Gồm 13 bộ dữ liệu amplicon gen 16S rRNA với độ đa dạng đầu vào từ 15 đến 59 loài vi khuẩn, tất cả đều nhắm mục tiêu vùng V4 của gen 16S rRNA.
Các chuỗi tham chiếu cho cả hai bộ dữ liệu được chuẩn hóa thành ASV-ref (chuỗi riêng biệt) và OTU-ref (chuỗi được gộp cụm) bằng mothur.
Tiền xử lý dữ liệu: Các bước tiền xử lý thống nhất được áp dụng để đảm bảo tính khách quan cho việc so sánh:
- Kiểm tra chất lượng chuỗi (FastQC).
- Cắt bỏ chuỗi mồi (cutPrimers).
- Gộp các đọc bắt cặp (paired-end reads) bằng USEARCH.
- Cắt độ dài chuỗi (PRINSEQ, FIGARO).
- Lọc bỏ các đọc bị định hướng sai (misoriented reads) bằng mothur.
- Lọc chất lượng bổ sung để loại bỏ các đọc chứa ký tự không rõ ràng hoặc có tỷ lệ lỗi cao (USEARCH).
- Để tách biệt hiệu ứng của việc gộp đọc với hiệu ứng khử nhiễu/gộp cụm, các đọc được phân tích theo hai kịch bản: đọc đơn (single-end – SE) và đọc bắt cặp đã gộp (paired-end – PE).
- Các mẫu mock được lấy mẫu lại thành 30.000 đọc mỗi mẫu để chuẩn hóa mức độ lỗi/nhiễu.
Các phương pháp được so sánh: Nghiên cứu so sánh hiệu suất của bốn phương pháp khử nhiễu ASV và bốn phương pháp gộp cụm OTU:
- Phương pháp khử nhiễu ASV: DADA2, Deblur, MED, UNOISE3.
- Phương pháp gộp cụm OTU: UPARSE, Average Neighborhood (AN), Opticlust, VSEARCH (DGC).
- Các thông số cụ thể cho mỗi thuật toán được đặt theo tài liệu bổ sung [13, Additional file 4].
- Các bước tạo OTU/ASV bao gồm huấn luyện mô hình lỗi (DADA2), lọc theo chế độ dương tính (Deblur), phân tách entropy (MED), và xác định chuỗi duy nhất để gộp cụm hoặc khử nhiễu (UPARSE, UNOISE3).
- Đối với các thuật toán của mothur (AN, DGC, Opticlust), chuỗi được sao chép, đếm, gộp cụm sơ bộ, và ma trận khoảng cách được tính toán, sau đó áp dụng ngưỡng khoảng cách 0.03 để gộp cụm.
- Loại bỏ chuỗi giả (chimera) được thống nhất bằng lệnh seq.error của mothur so với chuỗi tham chiếu của mock, và các đọc đơn (singleton reads) được giữ lại cho tất cả các phương pháp [15, Fig. 1].
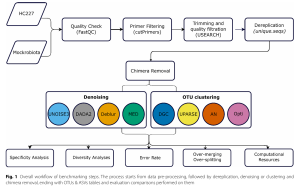
Phân tích và so sánh kết quả đầu ra: Các tiêu chí đánh giá bao gồm:
- Thành phần vi sinh vật, tỷ lệ lỗi, và định nghĩa ranh giới OTU/ASV.
- Phân tích đa dạng alpha: Chỉ số Shannon và Observed Feature (Sobs Calculator) bằng lệnh summary.single của mothur.
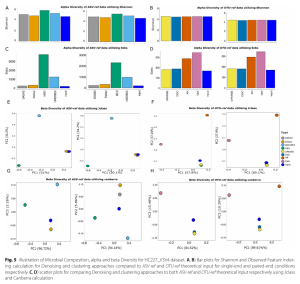
- Phân tích đa dạng beta: Khoảng cách giữa các phương pháp và ASV-ref/OTU-ref lý thuyết, sử dụng dist.shared và pcoa, với các độ đo khoảng cách như Jclass, Euclidean (tập trung vào sự hiện diện/vắng mặt) và Canberra (có xét cả độ phong phú).
- Đường cong Rarefaction và biểu đồ Alluvial để trực quan hóa đầu ra đa dạng.
Phân tích cụ thể:
- Phân tích độ đặc hiệu (Specificity analysis): OTU/ASV được phân loại thành khớp chính xác (100% giống chuỗi tham chiếu), không khớp (>97% giống), nhiễm bẩn (<97% giống nhưng >97% giống với SILVA), và các loại khác.
- Tính toán tỷ lệ lỗi (Error rate calculation): Xác định và loại bỏ chuỗi giả trước, sau đó so sánh OTU/ASV với chuỗi tham chiếu về lỗi không khớp (mismatches), chèn (insertions) và xóa (deletions).
- Phân tích quá gộp/quá phân tách (Merging/splitting analysis): Đánh giá các trường hợp nhiều chuỗi tham chiếu được gộp vào cùng một OTU/ASV (quá gộp) hoặc một chuỗi tham chiếu bị chia thành nhiều OTU/ASV (quá phân tách) bằng cách ánh xạ OTU/ASV với ASV-ref và OTU-ref.
- Phân tích chi phí tính toán (Computational cost analysis): Đánh giá thời gian chạy và mức tiêu thụ RAM với các bộ dữ liệu có kích thước khác nhau (5.000 đến 20.000 đọc). Cũng kiểm tra ảnh hưởng của các ngưỡng gộp cụm (0.01 đến 0.05).
Phân tích thống kê: Sử dụng kiểm định Shapiro-Wilk để đánh giá tính chuẩn của dữ liệu. Đối với dữ liệu chuẩn, áp dụng ANOVA và kiểm định HSD của Tukey; đối với dữ liệu không chuẩn, sử dụng kiểm định Kruskal-Wallis và kiểm định Wilcoxon rank-sum. Mức ý nghĩa 0.05.
Kết quả
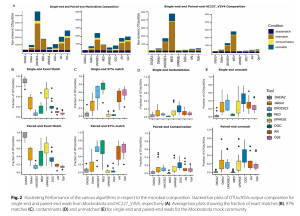
DADA2 và UPARSE cung cấp ước tính thành phần vi sinh vật chính xác nhất:
- Đối với bộ dữ liệu HC227_V3V4, DADA2 và UPARSE có số lượng khớp chính xác (exact matches) cao nhất, tiếp theo là DGC, AN, Opticlust và Deblur. Kết quả này nhất quán cho cả đọc đơn và đọc bắt cặp.
- MED và UNOISE3 có tỷ lệ không khớp (mismatches) cao nhất (75% và 61% cho HC227_V3V4).
- Về nhiễm bẩn (contaminants), DADA2, MED và UNOISE3 phát hiện khoảng 3% trong cộng đồng Mockrobiota, trong khi UPARSE và DGC phát hiện ít hơn.
- Hầu hết các phương pháp đều có khoảng 20 ± 4% đọc không khớp (unmatched reads), trừ DADA2 chỉ 5% cho HC227_V3V4.
DADA2 và UPARSE cho thấy tỷ lệ lỗi thấp nhất, nhưng vẫn có một số trường hợp quá gộp trong ASV-ref:
- DADA2, UPARSE và DGC thể hiện giá trị lỗi tổng thể thấp nhất, trong khi MED và UNOISE3 có tỷ lệ lỗi cao nhất cho cả hai bộ dữ liệu.
- Hiệu suất của Deblur không nhất quán giữa hai bộ dữ liệu.
- Khi so sánh với chuỗi tham chiếu ASV-ref, UPARSE và DGC cho thấy hiệu suất cao nhất trong việc gán OTU/ASV một cách chính xác, tiếp theo là DADA2.
- MED, UNOISE3 và Deblur xác định số lượng ASV được gán chính xác thấp nhất.
- UPARSE thể hiện sự quá phân tách (over-splitting) không đáng kể, tiếp theo là DGC và DADA2.
- Hầu hết các phương pháp đều bị quá gộp (over-merging) ở các mức độ khác nhau, trừ Opticlust và UNOISE3 có ít hoặc không có quá gộp. MED và DADA2 (đặc biệt là với đọc bắt cặp) cho thấy mức độ quá gộp cao hơn.
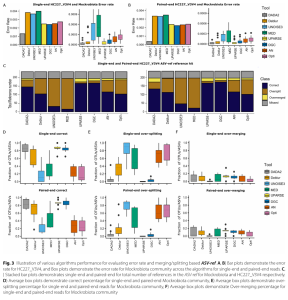
UPARSE, DGC và DADA2 có độ chính xác cao nhất với tỷ lệ quá phân tách và quá gộp thấp nhất trong OTU_ref:
- Khi so sánh với chuỗi tham chiếu OTU-ref (đã gộp cụm), UPARSE, DGC và DADA2 thể hiện tỷ lệ đọc được gán chính xác cao nhất và tỷ lệ quá phân tách thấp nhất.
- Các thuật toán AN, Opticlust và UNOISE3 chỉ có khoảng 20% chuỗi tham chiếu được khớp chính xác, cho thấy chúng có xu hướng phân tách các đọc tương tự từ cùng một tham chiếu.
- UNOISE3 và MED cho thấy tỷ lệ quá phân tách OTU/ASV cao nhất trong bộ dữ liệu HC227_V3V4.
- Điều đáng chú ý là 17% chuỗi tham chiếu HC227_V3V4 và 40% chuỗi tham chiếu Mockrobiota đã bị bỏ sót bởi tất cả các phương pháp thử nghiệm.
- Các trường hợp quá gộp ASV-references trong các thuật toán dựa trên gộp cụm đã được giải quyết hiệu quả khi sử dụng OTU-ref [34, Additional file 6].
- Ngưỡng gộp cụm 0.03 là tối ưu cho các thuật toán dựa trên gộp cụm, mang lại số lượng loài được gán chính xác cao nhất [35, Additional file 7].
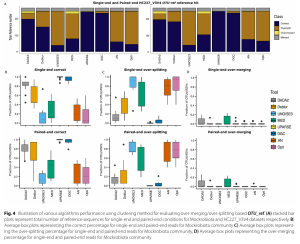
DADA2 và Deblur cho thấy sự tương đồng gần nhất với thành phần mock:
- Về chỉ số đa dạng alpha của Shannon, hầu hết các phương pháp đều rất giống với ASV-ref hoặc OTU-ref, trừ MED và UNOISE3.
- Đối với chỉ số Observed Feature, DADA2 và Deblur (không như MED và UNOISE3) cho thấy độ đa dạng vi sinh vật gần nhất với đầu vào.
- UPARSE cho thấy sự tương đồng gần nhất với thành phần mock (OTU-ref) trong các phương pháp gộp cụm.
- Đối với đa dạng beta, DADA2 và Deblur có sự tương đồng gần nhất với đầu vào ASV-ref, trong khi UPARSE thể hiện sự tương đồng gần nhất với OTU-ref.
- Biểu đồ Alluvial cho thấy DADA2 giữ lại một phần đáng kể sự đa dạng được tìm thấy trong ASV-ref, và UPARSE dường như duy trì một lượng lớn đa dạng của OTU-ref .
Tài nguyên tính toán và khám phá ảnh hưởng của tham số:
- Tất cả các thuật toán có thời gian thực hiện tăng dần theo số lượng chuỗi đầu vào. MED và Deblur yêu cầu thời gian chạy lâu hơn đáng kể (2 phút đến 120 phút) so với các thuật toán khác (2 giây đến 4 phút) [39, Additional file 9].
MED yêu cầu nhiều RAM nhất (lên đến 40 GB cho mẫu 20.000 đọc), trong khi các thuật toán khác yêu cầu từ 5 đến 11 GB RAM.
Thảo luận
Việc xác định ranh giới loài bằng trình tự amplicon 16S rRNA là một thách thức lớn. Nghiên cứu này giải quyết thách thức đó bằng cách sử dụng cộng đồng mock phức tạp nhất và các bước tiền xử lý, lọc thống nhất để so sánh khách quan các thuật toán dựa trên OTU và ASV.
- Tất cả các thuật toán đều có khả năng xác định các OTU/ASV không lỗi, nhưng chúng khác biệt rõ rệt trong việc xử lý các đọc sai. DADA2, UPARSE và DGC đạt hiệu suất cao nhất với số lượng OTU/ASV lỗi thấp nhất.
- MED và UNOISE3 có số lượng không khớp (mismatches) cao nhất.
- Hiệu suất của DADA2 giảm khi sử dụng các thông số mặc định thay vì các thông số thống nhất, tạo ra ít ASV khớp chính xác hơn và nhiều ASV không khớp hơn. Điều này cho thấy sự nhạy cảm của DADA2 với tiền xử lý và rằng các phương pháp ASV có thể xử lý/lọc nghiêm ngặt các chuỗi có độ phong phú thấp (như nhiễm bẩn và chimera).
- DADA2, UPARSE và Deblur cho thấy tỷ lệ lỗi thấp nhất, phù hợp với đánh giá chính xác của chúng về cộng đồng vi sinh vật. MED và UNOISE3 có tỷ lệ lỗi cao nhất và cho kết quả khác biệt đáng kể so với DADA2 và UPARSE, với số lượng loài vi khuẩn được gán chính xác thấp nhất và độ lệch lớn nhất so với dữ liệu đầu vào.
- Không có thuật toán nào có thể truy xuất đầy đủ số lượng chuỗi tham chiếu mock dự kiến (bỏ lỡ tới 17% cho HC227_V3V4 và 40% cho Mockrobiota). Vấn đề OTU inflation (ước tính quá mức độ phong phú của loài với nhiều dương tính giả) là một thách thức đối với hầu hết các thuật toán.
- UPARSE, DADA2 và DGC cho thấy độ chính xác cao nhất trong việc gán một ASV/OTU duy nhất cho mỗi chuỗi tham chiếu40. MED và UNOISE3 hoạt động kém. Các thuật toán dựa trên gộp cụm thường quá gộp các chuỗi tương tự, đặc biệt với ngưỡng ít chặt chẽ hơn [46, Additional file 7]. Các phương pháp dựa trên ASV như DADA2 và Deblur cũng cho thấy sự quá gộp và quá phân tách, điều này cho thấy cả hai vấn đề này đều tồn tại trong các quy trình ASV.
- DADA2 và UPARSE (và ở mức độ thấp hơn là Deblur) đạt được sự tương đồng gần nhất với tham chiếu lý thuyết về đa dạng vi sinh vật, được phản ánh qua các chỉ số đa dạng alpha và beta.
Mặc dù có sự khác biệt về độ phức tạp và vùng gen 16S rRNA được nhắm mục tiêu giữa các bộ dữ liệu mock, không có sự khác biệt lớn trong kết luận so sánh tổng thể. Tuy nhiên, cần lưu ý rằng các kết quả này chỉ dựa trên dữ liệu Illumina nhắm mục tiêu vùng V4/V3-V4 của gen 16S rRNA, và có thể khác biệt khi sử dụng các vùng gen khác hoặc các nền tảng trình tự khác.
Kết luận
Tóm lại, cả hai cách tiếp cận OTU và ASV đều tạo ra các kết quả khác nhau với ưu và nhược điểm riêng.
- Các thuật toán ASV tạo ra các biến thể chuỗi nhất quán, phù hợp cho các nghiên cứu mẫu độc lập hoặc phân tích tổng hợp mà không cần gộp cụm lại. DADA2 hoạt động tốt nhất trong số các phương pháp ASV, giữ được độ đa dạng ban đầu với ít sự gia tăng quá gộp, mặc dù việc sử dụng các thông số mặc định của nó dẫn đến quá phân tách và không khớp cao hơn đáng kể.
- Đối với các thuật toán dựa trên OTU, UPARSE hoạt động tốt nhất, với tỷ lệ gộp/phân tách cân bằng, có khả năng xử lý lỗi trình tự/chuỗi giả. UPARSE phù hợp cho các niche ít được nghiên cứu hoặc khi dự kiến có sự thay đổi lớn về vi sinh vật.
- Nhìn chung, DADA2 và UPARSE mang lại sự tương đồng gần nhất với cộng đồng vi sinh vật dự kiến với tỷ lệ lỗi thấp nhất và ít chuỗi giả nhất.
LOBI Vietnam là công ty tiên phong trong lĩnh vực Đọc trình tự gen thế hệ mới NGS (Next Generation Sequencing) và Phân tích Tin sinh học. Liên hệ hotline/Zalo 092.510.8899 để biết thêm chi tiết.
